蛋白质对每一个细胞活动都是至关重要的,解开它们的序列和结构是完全了解它们生物学的关键一步。早期的蛋白质测序方法主要是基于肽链的酶解或化学降解。随着人类基因组计划的完成和每个蛋白质可用信息的扩展,各种包含该序列信息的数据库形成了。
医学领域的进步是研究人员对人类生理学和各种相关生物过程的探索精神的结果。在分子水平上,蛋白质是重要生理功能所必需的四种重要大分子之一,由独特的氨基酸序列组成。
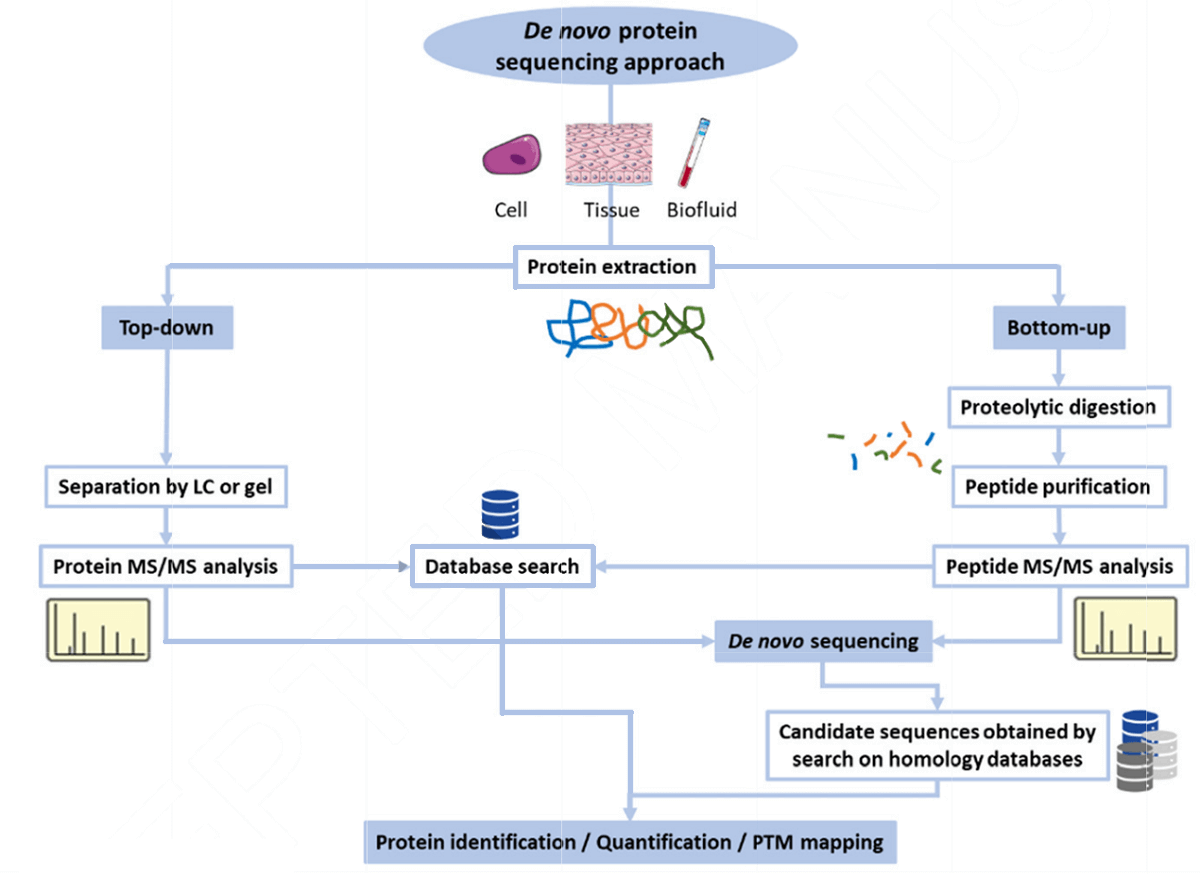
目前,尽管高分辨率质谱仪的可用性和高通量的发展对于新翻译后修饰(PTMs)的识别、翻译错误的注释或由于没有参考序列而不进行数据库搜索的鉴定,从头测序仍然是一个必要性的过程。对于药物设计中涉及的新蛋白和肽序列,从头肽测序仍然是首选的方法。新肽测序也有助于研究突变或PTMs产生的新蛋白形式。denovo测序的多种方法包括Edman降解、MS和阶梯测序。虽然Edman降解是时间密集型,但基于质谱的技术是经济和有效的。从本质上讲,通过MS进行蛋白质鉴定有两种方法,即自上而下和自下而上的蛋白质组学方法。在自下而上的方法中,蛋白质首先被消化,然后使用MS分析识别,而在自上而下的方法中,分析完整的蛋白质。在前者中,由于蛋白质的水解,样品的复杂性增加。多肽是用酶(如胰蛋白酶)直接在溶液中消化或用凝胶电泳方法分离后消化的。然后,在质谱分析之前,对消化的蛋白质进行电离。自下而上的方法允许对蛋白质进行量化,并提供PTMs位置的信息。在后者中,蛋白质序列信息包括PTMs、截断和序列变异,根据质量分析仪中使用的片段被保存下来。由于不稳定PTMs的测序在识别和序列定位方面存在挑战,ETD和高能碰撞解离(HCD)确保了更好的蛋白PTMs分析性能。在这些最初的蛋白质测序尝试之后,使用了散弹蛋白质组学技术。全基因组序列组合也用于基于同源性搜索的蛋白质测序。采用自下而上和自上而下方法相结合的各种策略进行从头测序。收集的MS/MS谱作为一个条形图,其中每个片段离子形成与其各自的m/z比相对应的峰。MS/MS谱图由n端肽片段对应的b离子和c端肽片段对应的y离子组成。在从头测序中,肽测序过程中产生的峰与20个标准氨基酸的参考峰进行比较。两个连续峰的相应质量的差异就产生了一个氨基酸的质量,这在原则上可以导致肽序列的确定。质谱还显示了由于样品内在变化或变化而发生在肽中的修饰处理。虽然可以用MS/MS谱预测序列,但一些峰可能会缺失; 因此,提出了使用特定算法进行快速序列预测的方法。
Aimsmass公司通过采取专业的分析软件和经验丰富的专业技术服务团队,积累了丰富的测序经验,能够提供基于质谱的蛋白或多肽的从头测序服务,获得100% 覆盖率和100% 准确性的氨基酸序列及氨基酸突变等信息,可弥补传统蛋白质鉴定方法在未知蛋白质序列和突变分析上的不足。
服务流程 |
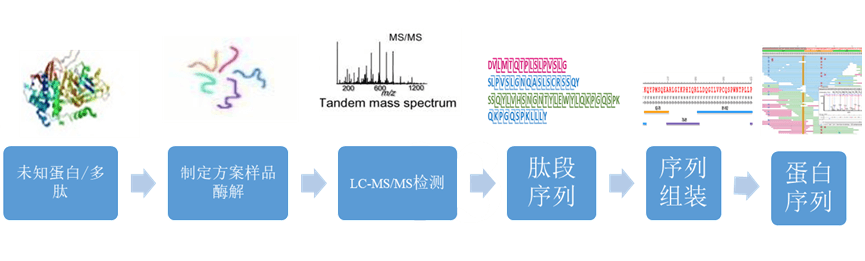
|
服务流程 |
|


